
Table of Contents
The Drug Development Process
Once a compound (Drug candidate) is selected and patented, so as to secure space for innovation within the fast moving research arena, the compound has to undergo several stages of preclinical (animal) testing and clinical (human) testing before it can make its way into the market. This post will provide an overview of the Drug Development Process.
The idea behind this is several folds. Until the discovery stage we only know that a compound has therapeutic value. What we don’t know is whether it is safe enough and effective enough to be used by humans. Also we need to work out what dose of the compound and in what form it needs to be given if it is to be brought into the market.
The process that a drug candidate undergoes during the development process makes sure that the drug is safe, effective and its dose and form are known. Also it ascertains that not only is the drug effective and safe on a set of humans that it has been tested on but also on a larger heterogeneous population.
The process of drug development is divided into the following steps:
- Pre-Clinical and Medical Development
- Phase I
- Phase II
- Phase III
- Pre-Registration
- Registration
- Phase IV
Figure below shows the various phases of Drug Development
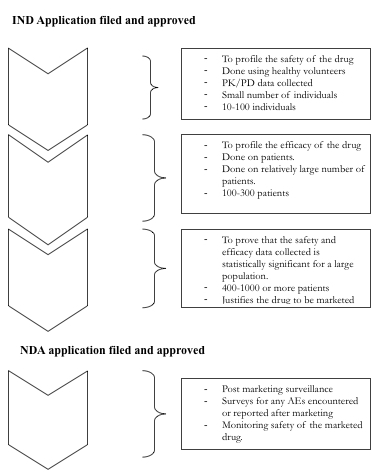
In this chapter we shall discuss about each of these steps.
PRE-CLINICAL AND MEDICAL DEVELOPMENT
The preclinical phase of testing of the drug candidate is to provide documentary evidence that it is fit for testing on humans. This is done by testing the drug on animals to explore its toxicity in every way that could harm a volunteer during the clinical phase.
The regulatory authorities such as the Food and Drug Administration (FDA) have stipulated tests for this, but further tests could be conducted are the need arises.
After the preclinical tests are conducted all the documented evidence is submitted to the regulatory authorities for approval to conduct further testing on humans. Such an application submitted to the regulatory authorities is called an Investigational New Drug (IND) application. Once the application has been approved the candidate can then be referred to as the investigational new drug and its testing on humans can begin. Starting preclinical development ascertains 10% success while successfully completing it and getting an IND approval ascertains 20% of the success of the trial.
PHASE I
Once the IND has been approved the next step is the Phase I. This phase involves testing the drug on humans for the first time to test its safety.
In this phase very small amount of drug is administered to healthy volunteers to understand the dynamics of the drug (such as does it go to the target? Or does it get concentrated in non-target areas within the body and what is the half life of the drug?). Also it is intended to study the kinetics of the body in response to the drug (such as any unexpected developments or side effects or toxicity levels of the drug).
These studies of what the drug does to the body (Pharmacodynamics) and what is the reaction of the body to the drug (Pharmacokinetics) in a healthy volunteer, are important for judging whether the drug is safe, tolerable, or changes need to be made in the dose, administration etc.
Though some Phase I studies yield positive results, as they are non-toxic and easily accepted by the body, in some cases even showing effectiveness in the Phase I itself. However in most cases the drug results in toxicity and safety concerns. If Phase I happens to go smoothly it ascertains that the drug tested in humans is safe and ascertains 30% success of the trial.
PHASE II
Once the safety of the drug is tested in Phase I, the next phase of development begins called the Phase II. This phase involves testing the drug for its efficacy by testing it on patients. This is the phase which basically decides where the study is heading as it is really important for a drug to be effective in patients. Successfully completing this phase ascertains 60% success in the study.
The results of this phase are also compared with those drugs already in the market, and to be competitive, any drug should be highly efficacious for the disease in question.
The phase II, involves testing the drug on relatively larger no of individuals, this time patients to test its effectiveness.
Phase II is an important phase considering that the drug is being tested for the first time in the actual patients for whom the drug is finally intended and if its data doesn’t turn out to be effective, there is no point of continuing the trial .
Phase II is also risky in the sense that the drug is being tested in patients and their underlying disease condition may cause the drug to behave in a different way that it did for healthy volunteers in Phase I, hence leading to Adverse Drug Reactions and complications.
Thus the patients must be monitored closely during this phase.
PHASE III
Once the safety and efficacy of the drug are known in Phase I and II respectively, the drug needs to be tested on a large heterogeneous population so that its tolerability and effectiveness can be judged, when it is used in a vide variety of large number of patients.
This phase is more of commercialization than the last phase of development. Phase III is important because the regulatory authorities need proof of the fact that the drug can be used by a large population of patients, without there being any adverse reaction or clinical significant findings. In other words the safety and efficacy profile of the drug should be statistically significant.
Phase II when completed successfully signifies there are 60% chances that the drug would reach the market. Phase III, therefore in mostly about consumers and regulators and to make them feel that it is all right to use the product universally.
Since the comparator arms of the study need to be large and also statistical significance need to be achieved, a large number of patients need to be recruited, thus making the Phase III a costly phase in drug development, and is costlier than phase II. Also phase III involves “head to head” studies, which involve giving the investigational new drug to one arm of the study and the comparator (generally an alternative drug regimen) to the other arm. Thus the new drug can be compared to its competitors, head to head, that is with a high degree of statistical confidence.
PRE-REGISTRATION
After the Last Patient Last Visit (LPLV) is completed in Phase III, An analysis of the data gathered during the three phases need to be done and write ups prepared accordingly.
This analysis along with the preclinical documentation is the submitted to the regulatory authorities. This application is called the New Drug Application (NDA). This process is important and requires taking care of details within submission and also preparing for certain questions which could be demanded by the regulatory agency.
Initially when pure paper based methods were uses, submissions used to be by submitting paper documentation, truckloads of paper for each submission, not to mention that it had to then be analyzed by the regulatory authorities. With the coming of electronic methods in data management, today submissions have become electronic which require integrated software and statistical packages such as SAS.
The two general principles that apply to clinical trials as a whole and specifically to submissions are:
- If it isn’t documented, it was never done.
- All data submitted should be traceable to its source
REGISTRATION
The registration process and timelines vary from country to country. In the US the Food and Drug Administration (FDA) is the regulatory agency and submits or rejects an application within ninety days from filing.
The FDA however prioritizes the review process based on the disease it serves, and the importance for a medication for that disease at that time. The FDA studies the submission for 6 to 12 weeks after which forms a committee of physicians who are renowned in that field of target disease. This committee then receives a presentation from the FDA as well as the company that has filed the application. There is then a vote of approval, to which the FDA give a lot of importance and in most probability keep their decision.
Before approving the drug the FDA also visits the manufacturing plants where the drug is to be manufactured. Once they are satisfied with everything, they may issue an approvable letter which may state that the drug is approvable subject to certain changes which they will recommend. Sometimes the FDA may issue an approvable letter while also stating that additional trials need to be done.
Thus there will be a time lag between when the approvable letter is issued and when the drug is actually approved for manufacturing. This timeframe may vary based on what changes/additional work/re-work the FDA requires. This was the case until 2002.
From 2004 onwards FDA has adopted a “complete letter process”, which means that it will classify letters of re-submissions into two types.
- Class 1 re-submissions would require minor changes that could be completed in a few months
- Class 2 re-submissions would require a larger timeframe.
PHASE IV
Phase IV trials are also called post-market surveillance studies. Once the approval is granted by the regulatory authorities, the drug is manufactured and marketed. Once it is marketed, patients are prescribed these medicines by doctors and consumed by the patients. Phase IV intends to monitor adverse effects of the drug on patients consuming the medication. The report of how the drug is doing in the market in terms of its safety and efficacy has to be reported to the regulatory authorities.
Also these studies are head to head studies as some patients are taking competitor drug and some the drug that has just been introduced, hence proving which drug is superior.
Also the Phase IV is sometimes necessary, as it can lead to optimization of dosing regimens, or test an investigator hypothesis and keep a check on difference in outcomes in the consumer.
The cost of developing drugs has been increasing over the years as can be seen in Figure below:
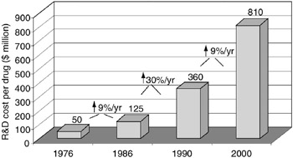
Thus the challenge here is to reduce R&D spending while increasing the number of NCEs (New Chemical Entities) discovered. This however is not the case. The number of NCEs being discovered has not shown much increase though R&D spending is increasing continuously. This is visible from an analysis in Figure below:
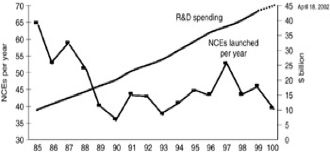
The pharmaceutical development process is quite risky and the benefits/risk ratio is variable. Sometimes a drug may be a blockbuster and some times it might just never make it to the market. The ideal situation expected is to reduce the cost of development, and yet not compromise on innovation.
This completes our discussion on Drug Development Process. We hope this post has helped you get insight into the Drug Development Process.
For a deep insight into the world of pharmaceutical research and development, subscribe to our Clinical Research Knowledgebase
Want a explore a career in Pharmaceutical Research and Development? Join our Diploma in Clinical Research program and kick-start a career in Clinical Research.
You may be interested in…
-
 eLearning
eLearningIntroduction to Clinical Research
$199.00 -
 eLearning
eLearningEpidemiology and Evidence-Based Medicine
$199.00 -
 eLearning
eLearningPharmaceutical Medicine
$299.00 -
 eLearning
eLearningEthics in Clinical Research
$199.00 -
 eLearning
eLearningRoles and Responsibilities in Clinical Research
$199.00 -
 eLearning
eLearningClinical Trial Preparation
$99.00 -
 eLearning
eLearningEssential Documents and Regulatory Submission
$199.00 -
 eLearning
eLearningClinical Trials Monitoring
$199.00